
Poradný výbor regulačného orgánu EÚ schvaľuje prípravu replikónu z Arcturus napriek vážnym bezpečnostným obavám. Konečné oficiálne schválenie je teraz na Európskej komisii, takže ide len o formalitu.
Odporúčam aj článok a možno konečne niečo pochopíte "tu čítaj" !!
Dňa 12. decembra 2024 Výbor pre lieky na humánne použitie (CHMP) (1.) vydal kladné stanovisko a odporučil povolenie na uvedenie lieku Kostaive na trh, čo je samoreplikujúca sa (replikovaná) injekcia mRNA vyvinutá spoločnosťou Arcturus Therapeutics .
Tu je vizuálna reprezentácia toho, ako tieto nebezpečné génové injekcie fungujú:
Japonsko schválilo tieto injekcie minulý rok . V novembri 2023 japonské ministerstvo zdravotníctva, práce a sociálnych vecí (MHLW) plne schválilo replikónovú vakcínu CSL a Kostaive ARCT-154 od spoločnosti Arcturus Therapeutics . Napriek obrovským obavám o bezpečnosť schválila japonská MHLW v septembri 2024 aktualizovanú posilňovaciu vakcináciu zameranú na líniu JN.1 subvariantov Omicron .
Tu čítaj 9.12.2023 Historické schválenie prvej celosvetovej vakcíny COVID-19 s vlastným zosilňovaním RNA (sa-mRNA) a tu 1.3.2024 VAROVANIE: Samorozširujúce sa vakcíny sú bližšie, než si myslíte!
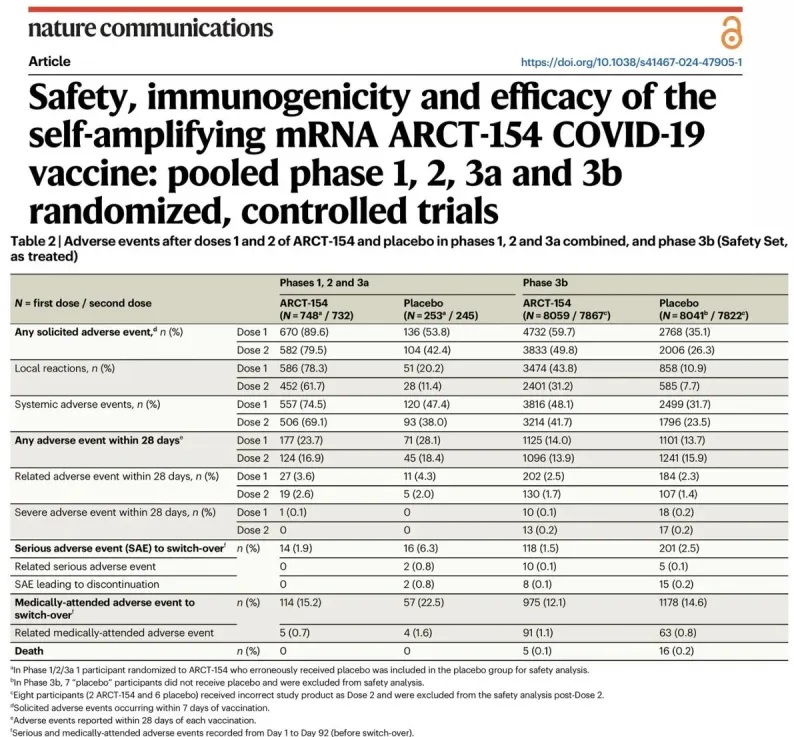
Počas klinických skúšok Kostaive zdokumentovaných (2.) bolo hlásených päť úmrtí medzi účastníkmi štúdie fázy 3b. V štúdiách vo fázach 1, 2 a 3a sa u 90 % injekčných účastníkov vyskytli nežiaduce udalosti, pričom 74,5 % hlásilo systémové reakcie a 15,2 % vyžadovalo lekársku pomoc po prvej dávke . Je pozoruhodné, že mnohí z autorov štúdie sú zamestnancami Arcturus Therapeutics na plný úväzok, čo vyvoláva obavy zo zaujatosti vo svojich záveroch.
Ako som vysvetlil "tu čítaj" , samoamplifikačný mRNA útok Biofarmaceutického komplexu sa už začal, pričom najmenej 33 kandidátov je už vo vývoji.
Tieto produkty sa správajú ako syntetický vírus. mRNA replikónu kóduje nielen cieľový antigén, ale aj vírusovú replikázu, takže mRNA sa môže replikovať v cieľových bunkách. Tento replikačný mechanizmus umožňuje neznáme trvanie produkcie toxického antigénu. Znepokojujúce je, že žiadna z klinických štúdií neriešila hlavný problém vypadávania.
Schválenie experimentálnych vakcín sa v Japonsku stretlo s rozsiahlym odporom vedcov a medicínskych expertov, ktorí už bili na poplach ohľadom „tradičných“ injekcií mRNA. V reakcii na blížiace sa zavedenie nových vakcín viedol japonský poslanec Ryuhei Kawada núdzovú tlačovú konferenciu v Japonsku, aby varoval verejnosť.
Replikóny sú ďalším logickým krokom k presunu výroby vakcín z bioreaktora v továrni do ľudského tela. Ako bolo uvedené , niektoré obchody odmietajú slúžiť očkovaným ľuďom. Dôvodom sú obavy z korónovej vakcíny ARCT-154 a jej schopnosti prenosu na iných ľudí.
Vzhľadom na obrovské objednávky zadané predsedníčkou Komisie Leyenovou na prvé prípravky genetického inžinierstva, ktoré zahŕňajú miliardy peňazí z našich daní, musíme predpokladať okamžité schválenie. Ďalšie objednávky nových, ešte nebezpečnejších preparátov by neboli prekvapením.
(1.) Z EMA https://www.ema.europa.eu/en/
Stanovisko EMA vydala stanovisko k tomuto lieku
Názov lieku
Kostaive
Dňa 12. decembra 2024 Výbor pre lieky na humánne použitie ( CHMP ) prijal kladné stanovisko, v ktorom odporučil udeliť povolenie na uvedenie lieku Kostaive na trh , vakcíny určenej na prevenciu COVID-19 u dospelých.
Žiadateľom o tento liek je Arcturus Therapeutics Europe BV
Kostaive bude dostupný ako prášok na disperziu na injekciu. Kostaive je vakcína proti COVID-19 založená na RNA (ATC kód: J07BN01). Obsahuje samozosilňujúcu sa mRNA, ktorá kóduje vrcholový proteín SARS-CoV-2. Samozosilnenie znamená, že mRNA tiež nesie pokyny na vytvorenie proteínu nazývaného replikáza. Po podaní do svalu vytvára replikázový proteín viac kópií mRNA, ktoré môže bunka použiť na vytvorenie väčšieho množstva vrcholového proteínu. Očkovanie s Kostaive indukuje produkciu neutralizujúcich protilátok a bunkovú imunitnú odpoveď zameranú na spike proteín, ktorý pomáha chrániť ľudí pred COVID-19.
Prínos lieku Kostaive ako základného očkovania proti COVID-19 sa ukázal vo veľkej štúdii, v ktorej dospelí dostali buď dve dávky lieku Kostaive alebo placebo. V porovnaní s placebom viedlo očkovanie Kostaive k zníženiu podielu pacientov, u ktorých sa vyvinul symptomatický COVID-19 medzi týždňom a 3 mesiacmi po druhej dávke vakcíny. Menšia imunopremostená štúdia tiež ukázala, že Kostaive je účinný ako heterológna posilňovacia vakcinácia (keď bola primárna vakcinácia vykonaná inou vakcínou proti COVID-19). Najčastejšie vedľajšie účinky lieku Kostaive sú reakcie v mieste vpichu (bolesť a citlivosť), artralgia, myalgia, bolesť hlavy, závrat, únava, triaška a horúčka.
Úplná indikácia je:
Kostaive je indikovaný na aktívnu imunizáciu na prevenciu COVID-19 spôsobeného SARS-CoV-2 u jedincov vo veku 18 rokov a starších.
Použitie tejto vakcíny má byť v súlade s oficiálnymi odporúčaniami.
Podrobné odporúčania na používanie tohto lieku budú opísané v súhrne charakteristických vlastností lieku (SmPC), ktorý bude uverejnený v Európskej verejnej hodnotiacej správe (EPAR) a bude dostupný vo všetkých úradných jazykoch Európskej únie po udelení povolenia na uvedenie na trh . zo strany Európskej komisie.
Podrobnosti o produkte
-
Názov lieku
-
Kostaive
-
Účinná látka
-
zapomeran
-
Medzinárodný nechránený názov (INN) alebo bežný názov
-
zapomeran
-
Terapeutická oblasť (MeSH)
-
Infekcia vírusom COVID-19
-
Anatomický terapeutický chemický (ATC) kód
-
J07BN01
-
Číslo produktu EMA
-
EMEA/H/C/006207
-
Žiadateľ o registráciu
-
Arcturus Therapeutics Europe BV
-
Stanovisko prijaté
-
12. 12. 2024
-
Stav názoru
-
Pozitívny
Správy o Kostaive
Účinnosť vakcíny (VE)
Vo fáze 3b bolo hlásených 3 632 (1 652 ARCT-154, 1980 placebo) prípadov podozrenia na COVID-19 od 1. do 92. dňa. Výtery z nosohltanu sa odobrali od 1920/3632 (52,9 %) prípadov do 3 dní, 978 (3632) 26,9 %) do 4–7 dní a 438/3632 (12,1 %) v priebehu 8–14 dní od nástupu symptómov. Nezávislý expertný výbor Event Adjudication Committee (EAC) posúdil 836 virologicky potvrdených prípadov a vyhodnotil 734 prípadov ako ochorenie COVID-19 (vrátane 48 prípadov závažného ochorenia COVID-19 a 10 úmrtí pripisovaných COVID-19) a 102 prípadov ako asymptomatického SARS- Infekcia CoV-2. Z týchto potvrdených prípadov COVID-19 sa 643 vyskytlo od 36. do 92. dňa, ale v troch prípadoch účastníci dostali vakcínu COVID-19 mimo štúdie, takže boli vylúčení z modifikovanej sady Intention to Treat (mITT) na analýzu, takže zostávalo 640 oprávnené prípady mITT (200 očkovaných a 440 príjemcov placeba), vrátane 43 závažných prípadov a 10 úmrtí pripisovaných COVID-19 (tabuľka 3 , Obr. 6 ). Cieľ primárnej účinnosti vakcíny (VE) bol splnený, pretože dve dávky ARCT-154 mali účinnosť 56,6 % (95 % CI: 48,7 – 63,3) proti ochoreniu COVID-19 akejkoľvek závažnosti s dolnou hranicou CI nad vopred špecifikovanou hranica úspešnosti 30 %. Sekundárne analýzy ukázali vysokú účinnosť proti závažnému COVID-19 (95,3 % [80,5–98,9]) a úmrtiu v dôsledku COVID-19 (86,5 % [-7,4–98,3]). Účinnosť proti akejkoľvek závažnosti COVID-19 bola podobná u mužských a ženských účastníkov (tabuľka 3 ). Účinnosť proti závažnému COVID-19 bola 100 % u zdravých 18 – 59-ročných a 91,9 % (37,9 – 98,9) u „rizikových“ účastníkov v rovnakej vekovej skupine. U dospelých vo veku 60 rokov alebo starších bola účinnosť 54,3 % (28,2–70,9) proti COVID-19 akejkoľvek závažnosti a 94,4 % (58,2–99,3) proti závažnému COVID-19.
Krivky kumulatívnej incidencie COVID-19 akejkoľvek závažnosti ( A ) a závažného COVID-19 ( B ) v skupinách s vakcínou a placebom od 36. dňa (podľa protokolu). Obrázok v plnej veľkosti
V sade mITT bolo analyzovaných 537 prípadov na identifikáciu zodpovedného variantu SARS-CoV-2, pričom 477 (88,8 %) bolo Delta (B.1.617.2) variant (164 a 313 v skupinách s vakcínou a placebom), dva boli Alfa ( oba placebo) jeden bol Beta (placebo) a dva boli varianty Omicron (jedna vakcína a placebo); vírusový variant nebol stanovený v 55 (10,2 %) prípadoch (doplnková tabuľka 5 ). Pri hodnotení iba v prípadoch mITT, v ktorých bola Delta (B.1.617.2) identifikovaným variantom, mali dve dávky ARCT-154 49,8 % (95 % CI: 39,3 – 58,4) účinnosť proti COVID-19 akejkoľvek závažnosti a 94,3 % ( 57,4–99,2) proti závažnému COVID-19 (doplnková tabuľka 6 ).
Ďalších 52 prípadov COVID-19 pozorovaných od 1. do 35. dňa, vrátane troch závažných prípadov, bolo posúdených ako vhodných na sekundárne analýzy účinnosti vo fáze 3b Intention to Treat (ITT), v ktorej bola účinnosť po akejkoľvek dávke ARCT- 154 od 1. do 92. dňa bola podobná mITT analýzam (tabuľka 3 , doplnkový obrázok 2 ); pozorovaná účinnosť proti COVID-19 akejkoľvek závažnosti bola 56,6 % (95 % IS: 49,0 – 63,1) a 95,6 % (81,5 – 98,9) proti závažnému COVID-19. Nakoniec vypočítaná účinnosť proti akejkoľvek závažnosti COVID-19 v združených účastníkoch fázy 1, 2 a 3a bola v súlade s pozorovaniami v štúdii fázy 3b ako účinnosť proti akejkoľvek závažnosti COVID-19 od 36. do 92. dňa (mITT) bola 56,3 % (95 % CI: 18,2 – 76,7) a od 1. do 92. dňa (ITT) bolo 58,9 % (95 % CI: 23,8–77,8) v tejto populácii (doplnková tabuľka 7 ).
Diskusia
Keďže sa stále objavujú nové varianty vírusu SARS-CoV-2, zistilo sa, že schválené mRNA vakcíny majú nižšie odhady účinnosti v porovnaní s mierami účinnosti nameranými v ich kľúčových štúdiách 6 , 7 , 8 . Dosiahli sme primárne ciele fáz 1, 2 a 3a tejto integrovanej štúdie, pričom sme úspešne preukázali prijateľnú bezpečnosť a reaktogenitu a imunogenicitu ARCT-154 s 95,9 % sérokonverziou na neutralizáciu protilátok proti variantu SARS-CoV-2 D614G. . Preukázanie bezpečnosti v týchto počiatočných fázach umožnilo nábor do väčšej populácie štúdie fázy 3b, v ktorej sme ukázali, že dve dávky ARCT-154 mali účinnosť vakcíny (VE) 56,6 % (95 % CI: 48,7 – 63,3) proti akejkoľvek závažnosti COVID-19 a najmä 95,3 % (80,5 – 98,9) proti závažnému COVID-19 na pozadí prevažne delta (B.1.617.2) variant SARS-CoV-2, ktorý spôsobil 88,8 % infekcií, kde bol tento variant identifikovaný. Toto je prvý publikovaný dôkaz klinickej účinnosti sa-mRNA vakcíny. ARCT-154 mal > 90 % účinnosť proti závažnému COVID-19 u osôb s rizikom závažného ochorenia, starších ako 60 rokov a dospelých vo veku od 18 do 59 rokov so základnými komorbiditami. Účinnosť proti úmrtiu v dôsledku COVID-19 bola 86,5 % (-7,4–98,3); široké medze spoľahlivosti odzrkadľujúce, že došlo iba k 10 úmrtiam, jednému očkovanému a deviatim príjemcom placeba.
Účinnosť prvých schválených mRNA vakcín bola preukázaná v čase, keď relatívne nízky podiel skúmanej populácie bol vystavený cirkulujúcemu vírusu SARS-CoV-2 a účinnosť bola meraná proti prototypu kmeňa Wuhan-Hu-1 alebo jedného z objavili sa prvé varianty, ktoré mali len malé zmeny v antigénnej štruktúre S proteínového cieľa týchto vakcín, skôr ako Delta (B.1.617.2). Pôvodné odhady účinnosti týchto vakcín proti ochoreniu COVID-19, vrátane závažného ochorenia, boli teda vyššie ako odhady pozorované pre ARCT-154, napr. 95 % (95 % CI: 90,3 – 97,6) pre BNT162b2 20 a 94,1 % (89,3-96,8) pre mRNA- 127321 . Pozorovalo sa však, že účinnosť týchto prvých schválených mRNA vakcín klesá proti vznikajúcim variantom, ktoré vyvolávajú obavy, čo sa zhoršuje oslabením imunity po počiatočnej sérii očkovania 5 , 6 , 7 , 8 . Ukázalo sa, že účinnosť dokončenej základnej očkovacej série schválených vakcín proti COVID-19 proti infekcii variantom Delta (B.1.617.2) sa pohybuje medzi 46 % a 91 % a od 47 % do 97 % proti závažnému COVID- 19 ochorenie spôsobené Delta (B.1.617.2) variant 22 .
Nižšia VE vakcíny ARCT-154, ktorú sme pozorovali, môže byť dôsledkom definície ochorenia COVID-19 použitej v štúdii, ktorá je založená na prítomnosti jediného symptómu v kombinácii s pozitívnou RT-PCR. To umožňuje zahrnúť do analýzy významný počet miernych a okrajovo symptomatických prípadov a uznáva sa, že účinnosť očkovania proti COVID-19 sa zvyšuje so závažnosťou COVID-19 a klesá s časom od očkovania. Účinnosť 3-dávkovej série vakcíny BNT162b2 proti miernemu, strednému a ťažkému COVID-19 spôsobenému variantom Omicron v priebehu 31 až 60 dní po poslednej vakcinácii bola 7,9 % (95 % IS: 2,3 – 13,1), 49,2 % (95 % CI: 46,8–51,4) a 76,4 % (95 % CI: 72,4–79,8), respektíve 23 . Bolo pozoruhodné, že ARCT-154 bol oveľa účinnejší proti závažnému COVID-19 ako ochorenie akejkoľvek závažnosti. Definícia prípadu použitá na analýzu primárnej účinnosti ako taká má významný vplyv na bodový odhad účinnosti. Klinické štúdie s inými vakcínami vo všeobecnosti používali „symptomatickejšiu“ definíciu ochorenia COVID-19 (pozitívna RT-PCR v kombinácii s najmenej dvoma systémovými symptómami alebo najmenej jedným respiračným znakom alebo symptómom).
Bezpečnosť a reaktogenita vo všetkých fázach naznačujú, že vakcína je dobre tolerovaná, prevažne s miernymi alebo stredne závažnými nežiaducimi účinkami, z ktorých väčšina boli prechodné lokálne reakcie. Najčastejšie vyžiadané systémové nežiaduce udalosti boli prechodná únava, myalgia, bolesť hlavy a triaška, ktoré ustúpili rýchlejšie ako lokálne reakcie. Všetky nežiaduce udalosti boli častejšie po očkovaní ako po placebe. Hoci štúdie neboli identické v dizajne, takže nie sú priamo porovnateľné, metódy hlásenia sú dostatočne podobné, aby sme mohli poznamenať, že celkové systémové AE a lokálne reakcie boli u príjemcov ARCT-154 podobné alebo menej časté ako licencované mRNA vakcíny24 . Hoci rozdiely v reaktogenite môžu súvisieť aj s kultúrnymi rozdielmi v hlásení subjektívnych nežiaducich udalostí pri použití posilňovacej dávky u dospelých s primárnou imunizáciou mRNA, ARCT-154 mal takmer identický profil reaktogenity ako vakcína mRNA, BNT162b2, keď bola pozorovaná na hlave. priame porovnanie dospelých v Japonsku 25 .
Hodnotenie imunogenicity očkovanej osoby proti kmeňu predkov SARS-CoV-2 pomocou testu sVNT ukázalo 94,1 % sérokonverziu u očkovaných štyri týždne po druhej dávke, čo bolo potvrdené pomocou validovaného mikroneutralizačného testu, ktorý ukázal 95,9 % sérokonverziu. V súčasnosti neexistuje žiadny sérologický korelát pre ochranu protilátkami anti-SARS-CoV-2, ale hladiny neutralizačných protilátok sú vysoko prediktívne pre imunitnú ochranu26 . Táto štúdia pravdepodobne predstavuje poslednú príležitosť na posúdenie účinnosti ARCT-154 v populácii naivnej SARS-CoV-2, keďže globálna pandémia viedla k tomu, že väčšina ľudí bola do určitej miery vystavená buď očkovaniu, prirodzenej infekcii alebo obom, čo vedie k hybridnej imunite 27 . Vzhľadom na vysokú úroveň pokrytia zaočkovanosťou s ubúdajúcou imunitou je súčasnou potrebou vakcín proti COVID-19 znovu nastoliť imunitu populácie na ochranu proti vznikajúcim variantom, ktoré stále spôsobujú prepuknutie 4 . Ako taký sa ARCT-154 s najväčšou pravdepodobnosťou použije ako posilňovacia dávka, a nie na primárnu imunizáciu, na zvýšenie a rozšírenie úrovne imunity proti cirkulujúcim variantom. Paralelná štúdia v Japonsku ukázala, že u dospelých plne imunizovaných mRNA vakcínami, hlavne BNT162b2, ako primárnou vakcínou, bola imunitná odpoveď na booster dávku ARCT-154 lepšia ako na booster dávku BNT162b2, keď sa merala ako neutralizačné protilátky. proti Wuhan-Hu-1 a podvariantu Omicron 4/5 25 . Ďalej, pretrvávanie odpovede na ARCT-154 bolo lepšie ako na BNT162b2 až 6 mesiacov po posilnení 28 . Licencované vakcíny boli teraz prispôsobené tak, aby odrážali meniacu sa epidemiológiu SARS-CoV-2 vrátane formulácií založených na S-proteíne najnovšieho variantu Omicron XBB.1.5, ktoré sú v súčasnosti odporúčanými vakcínami v Spojených štátoch 29 .
Predklinické štúdie na zvieratách s rovnakou patentovanou technológiou ako ARCT-154 ukázali, že poskytuje trvalú imunitnú odpoveď, ktorá zahŕňa indukciu neutralizujúcich protilátok a aktiváciu bunkami sprostredkovaných reakcií vrátane CD4+ T bunkového interferónu-γ a sekrécie interleukínu-4 antigén-špecifické CD8+ T-bunkové reakcie a pomer anti-spike proteínu IgG2: IgG1 indikujúci dominantnú odpoveď typu Th130 . Zostáva vidieť, či ARCT-154 indukuje podobné reakcie u ľudí. U dospelých vo veku 18-75 rokov Szubert et al. našli 1 μg a 10 μg dávky skúmanej lipidom zapuzdrenej SARS-CoV-2 sa-RNA vakcíny indukovanej neutralizačnými a anti-spike-IgG protilátkami, hoci medzi týmito dvoma meraniami bola len mierna korelácia 31 , 32 , 33 , 34 .
Štúdia bola nedávno dokončená a niektoré z obmedzení v tejto správe, ako sú 12-mesačné výsledky bezpečnosti a ďalšie skúmania imunogenicity, sa analyzujú a budú zverejnené v samostatných dokumentoch. Najmä dlhodobá bezpečnosť do jedného roka po očkovaní a ďalšie skúmanie humorálnej a bunkovej imunogenicity na objasnenie povahy imunitnej odpovede. Následné sledovanie tiež umožní posúdiť trvanlivosť tejto odpovede a hladiny krížovo neutralizujúcich protilátok proti novoobjaveným variantom. K dispozícii sú obmedzené údaje o skríženej neutralizácii, ale výsledky už spomínanej štúdie s posilňovacou dávkou v Japonsku ukazujú, že ARCT-154 vyvoláva lepšiu skríženú neutralizačnú reakciu proti Omicron BA.4/5 ako porovnávacia mRNA vakcína, a to tak v rozsahu 25 , ako aj v perzistencii 28 . Budú sa vyžadovať aj aktualizované formulácie založené na najnovších variantoch, ako napríklad Omicron XBB.1.5, ako už bolo uvedené. Ak sa-mRNA vakcíny poskytujú ekvivalentnú ochrannú účinnosť ako licencované mRNA vakcíny, ale s nižším množstvom mRNA, existuje potenciál na zníženie výrobných nákladov na dávku alebo na umožnenie výroby viacerých dávok, čo môžu byť dôležité faktory budúcej pandémie. 30 .
Táto prvá demonštrácia klinickej účinnosti sa-mRNA vakcíny ARCT-154 proti COVID-19 spolu s prijateľnou bezpečnosťou a reaktogenitou vo veľkej populácii štúdie stanovuje potenciál sa-mRNA vakcín pre budúce klinické použitie a dopĺňa ostatné štúdia, ktorá preukázala , že posilňovanie pomocou ARCT- 154 poskytuje lepšiu imunogenicitu proti Omicron ako vakcína mRNA25,28 . To podporuje ďalší vývoj sa-mRNA vakcín na rozšírenie armamentária proti budúcim prepuknutiam COVID-19.
Metódy
Protokol schválila etická komisia (EK) každého zo 16 študijných centier a Vietnamská národná komisia pre biomedicínsky výskum a ministerstvo zdravotníctva. Všetci účastníci poskytli informovaný súhlas pred zaradením do štúdie a štúdia bola vykonaná v súlade s etickými princípmi správnej klinickej praxe, Helsinskou deklaráciou a Medzinárodnou radou pre harmonizáciu (ICH E6R2) a platnými miestnymi regulačnými a bioetickými požiadavkami. Primárnymi cieľmi štúdií fázy 1, 2 a 3a bolo zhodnotiť v porovnaní s placebom bezpečnosť a reaktogenitu dvoch dávok ARCT-154 podaných s odstupom štyroch týždňov a imunogenicitu štyri týždne po druhej dávke. Primárnymi cieľmi štúdie fázy 3b bolo posúdiť bezpečnosť a reaktogenitu dvoch dávok ARCT-154 a ich účinnosť proti ochoreniu COVID-19 od 7 (36. deň) do 63 (92. deň) dní po druhej vakcinácii. Prieskumné hodnotenie účinnosti od 1. do 92. dňa sa uskutočnilo u všetkých účastníkov, ktorí dostali aspoň jednu dávku ARCT-154 v ktorejkoľvek fáze štúdie. Nezávislá komisia pre monitorovanie údajov a bezpečnosti (DSMB) mala plný dohľad nad štúdiou vrátane hodnotenia zaslepených údajov o bezpečnosti a potvrdených prípadoch COVID-19 a vydala odporúčania, aby sa pokračovalo v registrácii alebo v štúdii.
Účastníci a dizajn štúdie
Návrhy štúdií pre jednotlivé fázy štúdie sú znázornené na obr . Vo fáze 1 boli spôsobilými účastníkmi zdraví dospelí vo veku > 18 až < 60 rokov; vo fázach 2/3a/3b boli oprávnenými účastníkmi dospelí vo veku > 18 rokov. Dobrovoľníci zaradení do fázy 1/2/3a boli randomizovaní v pomere 3:1 a dobrovoľníci vo fáze 3b 1:1, aby dostávali ARCT-154 alebo placebo. Fáza 3b zahŕňala dobrovoľníkov so zvýšeným rizikom závažného ochorenia COVID-19 v dôsledku ich stavu komorbidity 31 alebo veku ≥ 60 rokov a randomizácia zahŕňala stratifikáciu týchto rizikových účastníkov. Účastníci fázy 1 boli prijatí a liečení ako prví, paralelný zápis pre fázy 2 a 3a bol povolený až po tom, čo DSMB a Ministerstvo zdravotníctva vo Vietname preskúmali všetky údaje o bezpečnosti zozbierané do 7 dní po druhej vakcinácii (36. deň) vo fáze 1. Podobne zápis pre fázu 3b bola schválená až po preskúmaní všetkých údajov o bezpečnosti zozbieraných do 7. dňa po prvej dávke vo fázach 2 a 3a.
Okrem vyššie uvedených vekových obmedzení boli vhodnými účastníkmi dospelí muži alebo ženy, ktorí mohli súhlasiť s účasťou, súhlasili s dodržiavaním všetkých požadovaných študijných návštev a postupov a boli ochotní poskytnúť požadované vzorky krvi a nazálnych výterov. Hlavnými vylučovacími kritériami boli dôkaz akútnej infekcie v čase zaradenia, tehotenstva alebo dojčenia, predchádzajúca infekcia COVID-19 (vrátane pozitívneho výsledku RT-PCR), blízky kontakt s osobou, o ktorej je známe, že je infikovaná SARS-CoV-2 alebo akákoľvek známa anamnéza anafylaktickej reakcie na vakcíny. Podrobné kritériá vylúčenia sú uvedené v doplnkovej tabuľke 2 .
Pri zaradení boli dobrovoľníci zaradení do skupín ARCT-154 alebo placeba pomocou systému interaktívnej odozvy (IRT), ktorý poskytoval jedinečný identifikačný kód štúdie a pridelený študijný zásah pre každého účastníka. Kódy boli prístupné len pre nezaslepený personál štúdie, ktorý pripravoval a podával vakcínu/placebo, ale nehral v štúdii žiadnu inú úlohu. Všetkým ostatným zamestnancom a účastníkom štúdie bolo zaslepené pridelenie štúdie. Na testovanie SARS-CoV-2 pomocou RT-PCR sa odobral tampón z nosa pri skríningu v 1. deň alebo do 5 dní od neho. V 1. deň po základnom odbere krvi na testovanie na protilátky špecifické pre nukleokapsidy SARS-CoV-2 a negatívne tehotenský test moču alebo krvi u žien vo fertilnom veku, bola podaná prvá priradená vakcína alebo injekcia placeba; druhá dávka bola podaná rovnakým spôsobom v deň 29. Aby sa zabezpečilo, že všetci účastníci dostali imunizáciu proti COVID-19, došlo k prechodu v deň 92, keď príjemcom placeba zo všetkých fáz bol ponúknutý ARCT-154 v dvoch dávkach s odstupom štyroch týždňov. Vakcíny z rôznych fáz dostali buď tretiu dávku ARCT-154 alebo dve dávky placeba. Táto správa prezentuje iba údaje získané do 92. dňa, údaje z prechodu budú prezentované samostatne.
Vakcína
ARCT-154 pozostáva z replikónu založeného na víruse venezuelskej konskej encefalitídy, v ktorom bola RNA kódujúca štrukturálne proteíny vírusu nahradená RNA kódujúcou spike (S) glykoproteín plnej dĺžky variantu SARS-CoV-2 D614G, zapuzdrený v lipidové nanočastice. 100 μg aktívnej zložky, skladovanej v liekovkách pri -20 °C alebo nižšej, sa rozpustilo v 10 ml sterilného fyziologického roztoku bezprostredne pred použitím a 0,5 ml dávky obsahujúce 5 μg sa podávali intramuskulárnou injekciou do deltového svalu. Placebo bol sterilný fyziologický roztok.
Bezpečnosť a reaktogenita
Po 30 minútach sledovania akýchkoľvek okamžitých reakcií všetci účastníci vyplnili elektronické alebo papierové študijné denníky počas 7 dní, počnúc dňom každej injekcie štúdie. Denníky si vyžiadali lokálne reakcie (erytém v mieste vpichu, bolesť, stvrdnutie/opuch a citlivosť) a systémové nežiaduce udalosti (AE; artralgia, triaška, hnačka, závrat, únava, bolesť hlavy, myalgia, nevoľnosť/vracanie a horúčka). Nevyžiadané AE boli zaznamenané až 28 dní po každej vakcinácii. Akákoľvek nežiaduca udalosť vedúca k prerušeniu alebo vylúčeniu zo štúdie, akákoľvek nežiaduca udalosť sprevádzaná lekárskym dohľadom (MAAE) alebo vážna nežiaduca udalosť (SAE) sa mala zdokumentovať počas jedného roka sledovania po dokončení počiatočnej série očkovania. Tu uvádzame všeobecné bezpečnostné údaje vrátane MAAE, SAES a stiahnutí do šiestich mesiacov (deň 210). Účastníci boli kontaktovaní prostredníctvom týždenných telefonátov, aby sa zabezpečilo dodržiavanie vyplnenia študijných denníkov, ktoré sa zbierali v dňoch 8 a 36, 7 dní po každom očkovaní a pri následnej návšteve v deň 57. Údaje o nežiaducich účinkoch boli zapísané do kazuistiky formou a príčinnú súvislosť udalostí stanovil vyšetrovateľ podávajúci správu.
Imunogenicita
Séra na analýzu imunogenicity sa odoberali v dňoch 1, 29 a 57. Primárnym cieľom imunogenicity bola odpoveď na 57. deň vo všetkých sérach dostupných od vhodných účastníkov fázy 1/2/3a, ako bolo merané vo Vietnamskom národnom inštitúte hygieny a epidemiológie laboratória použitím súpravu SARS-CoV-2 Surrogate Virus Neutralization Test (sVNT) (GenScript, Piscataway, NJ, USA). Táto súprava je funkčný enzýmový imunosorbentový test na kvalitatívnu alebo semikvantitatívnu detekciu protilátok (Nabs), ktoré blokujú väzbu SARS-CoV-2 na ľudský ACE2 receptor hostiteľských buniek. Protilátky boli vyjadrené ako skupinové geometrické priemerné koncentrácie (GMC), miery sérokonverzie (SCR) a geometrický priemer násobných vzostupov (GMFR) od 1. dňa. Výsledky boli vyjadrené v jednotkách na ml (U/ml) kalibrovaných so štandardným sérom WHO.
Na potvrdenie pozorovaní z prieskumného testu sa imunogenicita hodnotila aj vo validovanom mikroneutralizačnom teste založenom na bunkách 293T-ACE2 Bioanalytickým laboratóriom pre vývoj farmaceutických produktov (PPD, Richmond, VA, USA). Týmto sa merali neutralizačné protilátky proti SARS-CoV-2 v sére z 1. a 57. dňa od všetkých účastníkov fázy 1, prvých 150 vzoriek z fázy 2 a náhodne vybraných vzoriek od ostatných účastníkov vo fáze 2 a všetkých dostupných vzoriek z fázy 3a. .
Hodnotenie účastníkov s podozrením na COVID-19
Na účely hodnotenia účinnosti boli účastníci s podozrením na COVID-19 vyhodnotení na prítomnosť potenciálnych symptómov a klinických príznakov COVID-19 vrátane horúčky, zimnice, kašľa, dýchavičnosti alebo ťažkostí s dýchaním, únavy, bolesti svalov alebo tela, bolesti hlavy, novej straty chuti alebo vône, bolesť hrdla, upchatý nos alebo nádcha, nevoľnosť, vracanie alebo hnačka. Ktorýkoľvek z týchto príznakov, ktorý sa vyskytne po 3 dňoch po očkovaní, spustil diagnostické testovanie na COVID-19. Ak to bolo možné, účastníci navštívili svoju príslušnú študijnú kliniku, kde sa odobrali nosové výtery na RT-PCR s dokumentáciou o anamnéze a užívaných liekoch. Prípad COVID-19 definovaný protokolom musel mať virologické potvrdenie (pomocou RT-PCR) SARS-CoV-2 a aspoň jeden zo symptómov alebo klinických nálezov uvedených vyššie.
Definície prípadov pre hodnotenia COVID-19 a závažného COVID-19 boli založené na odporúčaniach americkej FDA v súlade s podobnými klinickými štúdiami, ktoré pre závažný COVID-19 zahŕňali ktorékoľvek z nasledujúcich: akútna pľúcna, srdcová, renálna, hepatálna alebo neurologická dysfunkcia ; šok; smrť; alebo prijatie na jednotku intenzívnej starostlivosti (doplnková tabuľka 3 ). Všetky prípady podozrenia na COVID-19 prešli zaslepeným odstupňovaným hodnotením nezávislým výborom na rozhodovanie udalostí (EAC) zloženým z klinických odborníkov so skúsenosťami v diagnostike, starostlivosti a liečbe COVID-19. EAC preskúmala zaslepené údaje z každého prípadu a dospela k záveru, či prípad spĺňa protokolom definované kritériá prípadu COVID-19 a závažnosť podľa klasifikácií americkej FDA a WHO. Do účinnosti primárnej vakcíny (VE) sú zahrnuté iba virologicky potvrdené, protokolom definované prípady posúdené EAC.
Štatistická analýza
Primárne bezpečnostné koncové body boli hodnotené v súbore analýzy bezpečnosti (SAS; všetci účastníci, ktorí dostali akúkoľvek injekciu štúdie) a súbore analýzy reaktogenity (RAS; všetci účastníci, ktorí dostali akúkoľvek injekciu štúdie a poskytli aspoň jednu správu v denníku). Štatistická analýza údajov o bezpečnosti a reaktogenite bola popisná s frekvenciou a percentom pre účastníkov analyzovaných podľa študijnej skupiny.
Analýza primárnej imunogenicity v súbore analýzy imunogenity (IAS) zahŕňala všetkých účastníkov, ktorí dostali obe priradené injekcie štúdie v hodnotenom časovom bode bez dôkazu predchádzajúcej infekcie SARS-CoV-2 v deň 1 (tj boli séronegatívni na N-protilátku) a v aspoň jeden platný výsledok testu imunogenicity po očkovaní. GMC sa vypočítali ako priemer log-transformovaných výsledkov a potom sa umocnil priemer (aby sa výsledky prezentovali na pôvodnej stupnici). GMFR sa vypočítal ako priemer rozdielu po logaritmicky transformovaných výsledkoch (po základnej hodnote mínus základná hodnota) a umocnení priemeru. Obojstranný 95 % CI pre GMC a GMFR sa získal logaritmickou transformáciou výsledkov protilátok; 95 % CI sa vypočítal na základe Studentovej t-distribúcie pre priemerný rozdiel, potom sa umocnili limity spoľahlivosti. Sérokonverzia bola definovaná ako 4-násobné zvýšenie titra oproti základnej hodnote a jej obojstranný 95 % CI bol vypočítaný použitím Clopper-Pearsonovej metódy.
Primárny cieľ účinnosti bol hodnotený v modifikovanej sade Intention to Treat (mITT) zloženej zo všetkých účastníkov, ktorí dostali obe priradené injekcie štúdie a nemali žiadny dôkaz infekcie SARS-CoV-2 v deň 1 a až do dňa 36, 7 dní po druhá študijná injekcia. Prvý primárny koncový ukazovateľ bol definovaný ako prvý výskyt potvrdeného, protokolom definovaného COVID-19 s nástupom medzi 36. a 92. dňom vrátane. Pre celkový cieľ primárnej účinnosti štúdie bola nulová hypotéza, že účinnosť vakcíny (VE) ARCT-154 na prevenciu COVID-19 bola ≤ 30 % (tj účinnosť H0 : VE ≤ 0,3). Účinnosť vakcíny sa vypočítala z pomeru rizika 1, kde pomer rizika (HR) a 95 % CI sa odhadujú pomocou Coxovej proporcionálnej regresie rizika. Primárny cieľ účinnosti by bol splnený, ak by spodná hranica 95 % CI pre VE presiahla 30 %; celkovo bolo potrebných 372 prípadov COVID-19 na poskytnutie približne 90 % energie na zistenie 50 % zníženia miery nebezpečenstva (50 % VE). Faktory použité ako kovariáty v Coxovej proporcionálnej regresii rizika zahŕňali: Riziková skupina: ≥ 18 až <60 rokov a „zdravý“, ≥ 18 a <60 rokov a „rizikový“ a ≥ 60 rokov a oblasť miesta štúdie. Ak bol splnený primárny cieľ účinnosti, podľa hierarchického prístupu sa testovala aj nulová hypotéza, že účinnosť vakcíny na prevenciu výskytu potvrdeného závažného ochorenia COVID-19 bola ≤ 0 % (tj účinnosť H0 : VE ťažká ≤ 0). Sekundárne hodnotenie účinnosti sa uskutočnilo v súbore ITT, ktorý zahŕňal všetkých účastníkov, ktorí dostali aspoň jednu injekciu štúdie, pričom sekundárnym koncovým bodom bol výskyt potvrdeného, protokolom definovaného COVID-19 s nástupom kedykoľvek po dávke 1 až do Deň 92 vrátane.
Súhrn prehľadov
Ďalšie informácie o dizajne výskumu sú k dispozícii v súhrne výkazov Nature Portfolio, ktorý je prepojený s týmto článkom.
Zdroj:
Petícia za vyhlásenie referenda
S prosbou. Financujeme sa výlučne prostredníctvom vašich darov. Veľká vďaka
Všetky práva vyhradené © OZ Dôstojnosť Slovenska.
